Last ned PDF:
Genetisk mangfold i Borrelia.pdf (208 KB)Bakgrunn
Borrelia burgdorferi sensu lato (s.l.) er en samlebetegnelse for flere Borrelia-arter, som på verdensbasis er assosiert med flått fra familien Ixodidae, herunder Ixodes ricinus (skogsflått). I Europa er Borrelia afzelii, Borrelia garinii og Borrelia burgdorferi sensu stricto (s.s.) ansett for å være de tre vanligste humanpatogene Borrelia-artene (1, 2). Borrelia-spiroketer er heliksformede bakterier med en lagdelt cellulær struktur og flageller - som er et karakteristisk trekk for spiroketer (2). I 1982 oppdaget Willy Burgdorfer Borrelia-spiroketer i flått, og Borreliose ble identifisert som en vektorbåret zoonose som kunne overføres til mennesker via flått (3). Ulike genotypingsmetoder har de siste årene vært tatt i bruk for å studere evolusjon og taksonomi av gruppen med Borrelia-spiroketer som utgjør B. burgdorferi s. l. Metoden multilokus sekvenstyping (MLST) har avslørt et stort genetisk mangfold hos ulike Borrelia-stammer, noe som har ført til taksonomiske endringer (4-10).
Borreliose kan i sjeldne tilfeller gi alvorlig sykdom. Symptomene er blant annet feber, ubehag, erythema migrans (EM), lyme artritt og nevrologiske lidelser (11). Human borrelioseinfeksjon kan deles i inntil tre faser med forskjellige kliniske manifestasjoner. I de tidlige stadiene av infeksjonen kan man få en lokal hudinfeksjon (trinn 1) som kan bli etterfulgt av spredning til hele kroppen (trinn 2). I tillegg indikerer forskning at noen pasienter har lange ettervirkninger (trinn 3) (12, 13).
Forskning indikerer også at B. afzelii og B. garinii kan gi forskjellige kliniske manifestasjoner, noe som kan tyde på et ulikt patogent potensial (4). B. afzelii er forbundet med mild systemisk infeksjon, EM og lymphocytoma i de tidlige stadiene, etterfulgt av acrodermatitis chronica atrophicans (ACA) som rammer det perifere nervesystemet. B. garinii kan forårsake EM og lymphocytoma i de tidlige stadiene, men er vanligvis forbundet med nevrologiske lidelser (11-13). B. burgdorferi s.s. gir sjelden EM i Europa, men opptrer noe annerledes i USA. B. burgdorferi s.s. kan føre til en sjelden tilstand kjent som Lyme Karditt (hjertebetennelse)(14).
Materiale og metoder
Denne artikkelen er basert på søk i PubMed (http://www.ncbi.nlm.nih.gov/pubmed/) og de globale MLST databasene (http://www.mlst.net/ og http://pubmlst.org/). Søkeord som ble brukt; «MLST, Borrelia, Genetics, Host, Taxonomy, Evolution, Lyme». Relevante artikler ble valgt på grunnlag av tittel, sammendrag, forfattere og tidsskriftet hvor artiklene er trykket. Forfattere ble valgt på bakgrunn av publikasjonsliste, forskningstilhørighet og fagområder de publiserte innenfor. De fleste forfatterne tilhører de store fagmiljøene som jobber med Borrelia-MLST på verdensbasis. Litteratursøket er gjennomført i forbindelse med doktorgradsavhandlingen med tittel «Tick-borne pathogens: Detection and characterization of Borrelia burgdorferi sensu stricto, Borrelia afzelii, Borrelia garinii and Borrelia valaisiana in Ixodes ricinus ticks» (15).
Taksonomi
Bakteriell taksonomi er en vitenskapelig disiplin som stadig utvikles og endres. Tidligere ble en ny bakterievariant inkludert i en art dersom DNA-DNA hybridisering viste mer enn 70 prosent likhet mellom den nye varianten og en kjent artsklassifisert stamme. Nye analysemetoder som MLST og DNA-sekvensering har gitt enklere måter å karakterisere nye stammer på, blant annet ved å måle genetisk distanse. Nye stammer kan verifiseres som nye arter eller som tilhørende en eksisterende art mye raskere enn tidligere, og dette påvirker utviklingen av den bakterielle taksonomien (8).
Borrelia-spiroketer ble opprinnelig delt i to hovedgrupper; de artene som forårsaker borreliose og de som forårsaker tilbakefallsfeber (infeksjon overført av lus eller flått som gir sepsis med tilbakevendende feberanfall, forekommer ikke i Norge). Den hovedgruppen som forårsaker borreliose inkluderer B. burgdorferi s.l., og i denne gruppen fins tre kjente humanpatogene arter, B. afzelii, B. garinii og B. burgdorferi s.s., og 15 mindre patogene eller ikke-patogene arter (per d.d.), blant annet Borrelia lusitaniae, Borrelia tanukii, Borrelia turdi, Borrelia spielmanii og Borrelia valaisiana. I nyere tid har B. garinii blitt delt inn i to grupper, en gruppe som i hovedsak infiserer fugler og en gruppe som i hovedsak infiserer smågnagere. Den siste gruppen har fått tildelt navnet Borrelia bavarensis, etter regionen Bayern (Bavaria) i Tyskland hvor den først ble først oppdaget (7).
Den andre hovedgruppen, som forårsaker tilbakefallsfeber, består av om lag 20 forskjellige arter, blant dem Borrelia duttoni og Borrelia hermsii (2). Nyere studier viser at Borrelia-spiroketer også kan deles inn etter hvilke vertsdyr de benytter seg av. B. afzelii og B. bavarensis er assosiert med smågnagere, mens B. garinii og B. valaisiana er assosiert med fugler. B. burgdorferi s. s. er beskrevet som en generalist og er en av Borrelia-spiroketene som kan benytte seg av mer enn én bestemt type vertsdyr (16).
Borreliagenomet
I 2002 ble det første fullsekvenserte genomet av Borrelia burgdorferi sensu stricto (stamme B31) publisert (17). Genomet består av et 910 725 basepar (%GC = 28,5) lineært kromosom samt 9 sirkulære og 12 lineære plasmider. Det er identifisert mer enn 150 lipoprotein-kodende gener, og studier av genomet har vist en høy mutasjonsfrekvens, samt evne til genetisk rearrangering (17). Borrelia-spiroketers naturlige levested er in vivo, enten i en vert eller i en vektor. Ved bruk av mikromatriser er det vist at enkelte gener i Borrelia-genomet uttrykkes ulikt i ulike miljøer. Studien ble utført på Borrelia-stammer isolert fra ulike levesteder, og forskjellen i genuttrykket ser ut til å være indusert av verten. Disse funnene tyder på at Borrelia-spiroketer lever mer som parasitter, med stor evne til å tilpasse seg ulike verter (1, 18).
I epidemiologistudier og studier av genetisk utvikling hos de forskjellige Borrelia-artene, har et utvalg av såkalte husholdningsgener (gener som koder for proteiner som er nødvendig for de normale cellefunksjonene) blitt sekvensert og karakterisert. Det samme er blitt gjort med ikke-kodende DNA-sekvenser og gener med høy mutasjonsfrekvens. Studiene viser at overflateproteinene representerer en gruppe av gener med høy mutasjonsfrekvens. De ytre overflateprotein (Osp)-genene er lokalisert i både lineære (OspA/B) og sirkulære plasmider (OspC). Studier av disse svært variable genene har gitt informasjon om genetisk homogenitet, og identifisert horisontal genoverføring mellom arter (8).
Det er også påvist at de ytre overflateproteinene uttrykkes ulikt i forskjellige miljøer. Uttrykket av OspA, OspB og OspC er koblet til spesifikke leveforhold, enten det er i pattedyr eller i flått. Yang et al. (2004) har vist at OspA og OspB spiller en viktig rolle i koloniseringen av Borrelia-spiroketer i den midtre delen av tarmen hos flåtten (19). Et utvalg av husholdningsgener har blitt brukt i molekylære metoder for å studere utbredelsen av spesifikke Borrelia-arter. Kromosomale gener som 16S rRNA, groEL, hbb, flaB, recA, clpA, clpX, nifS, pyrG, pepX, rplB, recG og uvrA, representerer et utvalg av konserverte gener med lav mutasjonsfrekvens. De fleste av disse genene har tilstrekkelig grad av heterogenitet mellom de ulike Borrelia-artene til at de kan benyttes i utviklingen av artsspesifikke deteksjonsmetoder (5, 9, 10). Studier har vist at 16S rRNA-regionen er svært konservert, og at det kan være utfordrende å etablere artsspesifikke deteksjonsmetoder i denne regionen. De andre nevnte genene er alle anvendt i MLST-metoder som brukes til å skaffe kunnskap om Borrelia-genomets utvikling (5-7, 9, 10).
Metoder for måling av evolusjon og genetisk diversitet
Genotypingsmetoder er basert på nukleotidsekvensen til ulike gener, og utgjør en pålitelig og reproduserbar analysemetodikk som kan påvise polymorfismer i utvalgte gener. Genotypingsmetoder kan være basert på kutting med restriksjonsenzymer, DNA-kopiering med PCR-metoder, DNA-sekvensering eller en kombinasjon av disse metodene (20).
Multilocus sekvenstyping (MLST).
MLST er en sekvensbasert karakterisering av bakterier og andre organismer. C. J. Maiden et al. (20) utviklet MLST basert på analyseprinsippet fra multilokus enzym elektroforese (MLEE). MLEE er blitt brukt til å beskrive genetisk variasjon hos husholdningsgener ved hjelp av restriksjonsenzymer. Metoden ga gode resultater, men det var vanskelig å sammenligne resultater fra forskjellige laboratorier (21, 22). Analyseprinsippet fra MLEE er overført til MLST, som er en mer reproduserbar metode. MLST kan brukes som et globalt epidemiologisk overvåkingsverktøy slik at forskjellige laboratorier kan sammenligne genotypingdata (22). Det er utviklet en online database for MLST genotypingsdata, og metoden regnes som nøyaktig og svært detaljert (23).
MLST kan brukes til å beskrive bakteriestammer basert på genetisk variasjon. Metoden kombinerer PCR-amplifisering med sekvensering av fragmenter fra valgte husholdningsgener og bioinformatiske analyser (figur 1). Bioinformatikk kan brukes til å koble informasjon om global epidemiologi og lokalt mangfold mellom ulike bakteriestammer. Bioinformatikk kan også brukes til å fastslå om det finnes sporbare genetiske relasjoner (22, 24, 25). MLST, slik som Urwin og Maiden (25) beskrev den, har strenge kriterier for hvilke gener som er egnet til MLST genotyping. De bør være basert på husholdningsgener som har moderat mutasjonsrate, er cirka 450 - 500 basepar i størrelse og det bør velges gener fra hele genomet for å unngå lokale systematiske feil. De valgte genene bør ikke være flankert av gener som er under sterkt seleksjonspress, og de bør ha et jevnt genetisk mangfold for å bidra likt til den genetiske analysen (25). I MLST gis alleler fra de undersøkte genene et nummer basert på sekvensinformasjon, og videre analyser er basert på allelnummer og sekvenstype. MLST bruker klyngeanalyse for å beskrive slektskapsrelasjoner mellom de ulike allelene. Metoden er nyttig for identifikasjon og klassifisering av stammer med kjent artstilhørighet, men gir ufullstendig fylogeni (slektskapstre) og er ikke egnet for å definere nye arter (22, 25).
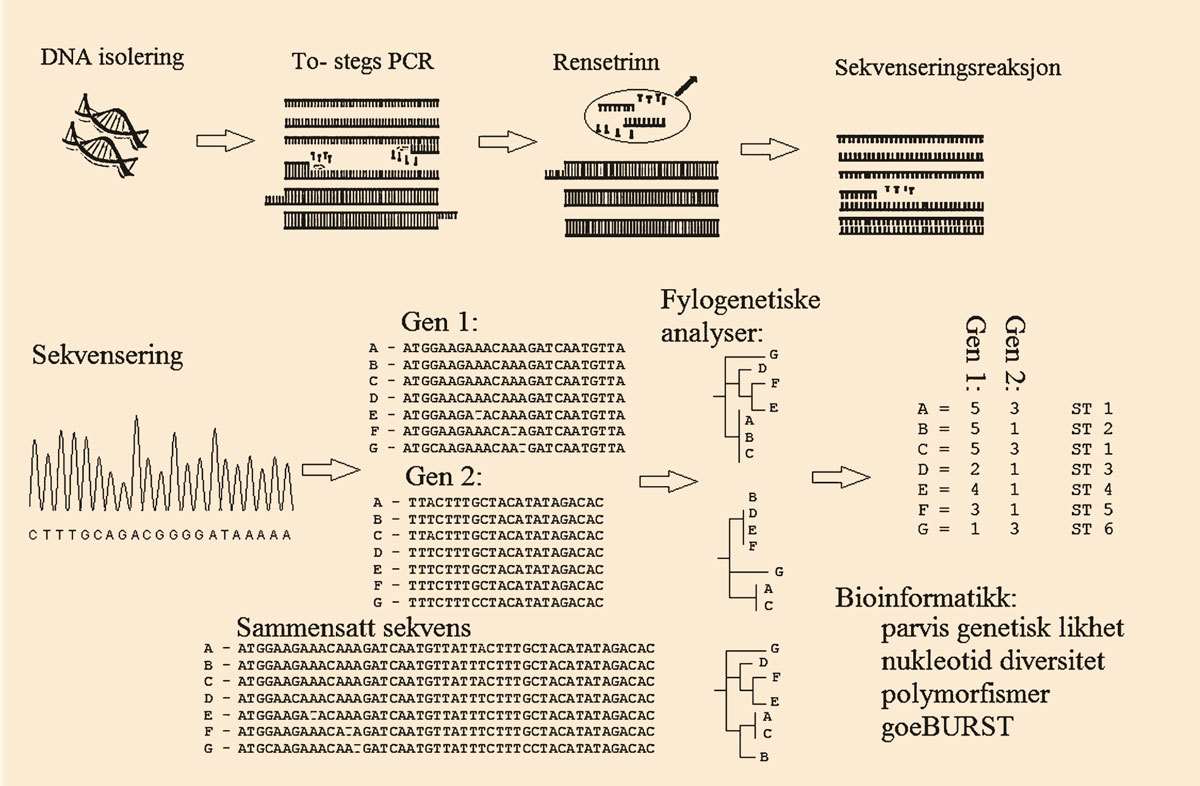
Multilokus sekvens analyse (MLSA)
MLSA, en alternativ bakteriell genotyping, benytter samme analyseprinsipp som MLST. MLSA er også nukleotidsekvensbasert med sekvenser fra flere gener - ofte de samme genene som brukes i MLST. Til forskjell fra MLST bruker MLSA selve sekvensen i videre analyser, og ikke et allelnummer. MLSA benytter avstandsmetode og parvis genetiske likheter for å beskrive slektskapsrelasjoner. Dette gir et mer nyansert bilde av den genetiske utviklingen (9). Den parvise genetiske likheten kan brukes til å bestemme en grenseverdi for artstilhørighet. Terskelverdien for B. burgdorferi s.l. ble beregnet til < 0,017 (1,7 % genetisk ulikhet) innen de ulike Borrelia-artene. Dette har gjort det mulig å bruke MLSA til å definere nye arter og taksonomisk tilhørighet, basert på likheter og ulikheter mellom Borrelia-stammer (5). Metoden har sine begrensinger, og de kriteriene som ble utarbeidet av Urwin og Maiden (25) blir ikke alltid fulgt når det etableres nye MLST-metoder.
Utvikling av genotypingsmetoder
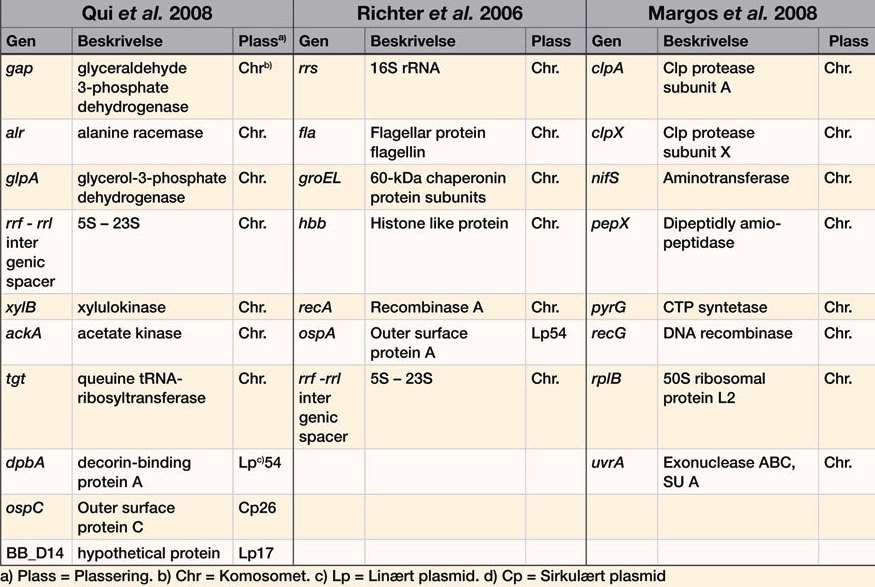
Både MLST og MLSA har vært benyttet i populasjonsstudier og slektskapsanalyser av Borrelia-arter fra forskjellige geografiske områder. Metodene er brukt både til taksonomi- studier og karakterisering av nye Borrelia-arter (5-7, 9, 10). I 2006 publiserte Richter et al. (9) en MLSA-metode for karakterisering av B. burgdorferi s. l., basert på fem husholdningsgener samt en intergenic spacer region (IGS) og ospA som er plassert på et lineært plasmid, og i 2008 publiserte Margos et al. (5) en MLST-metode basert på åtte husholdningsgener for taksonomi, populasjon og evolusjonære studier (tabell 1). Metoden har blitt videreført som MLSA-metode i andre studier av Margos et al. (6, 7, 26). Samtidig, i 2008, presenterte Qiu et al. (10) en MLST metode med en kombinasjon av seks husholdningsgener, IGS og plasmidgener under positiv seleksjon (tabell 1). Bare MLST-metoden av Margos et al. (2008) inkluderer husholdningsgener som oppfyller de strenge kriteriene som Urwin og Maiden (25) la til grunn for metodens validitet. Mens de andre MLST- og MLSA-metodene kombinerer plasmidgener, husholdningsgener og ikke-kodende gener, er MLST-metoden utviklet av Margos et al. (5), kun basert på husholdningsgener med fragmentstørrelser mellom 564 - 651 basepar og et jevnt nivå på det genetiske mangfoldet.
MLST er tidkrevende, og siden kostnadene for fullgenomsekvensering (FGS) er på vei ned, bruker forskere stadig oftere FGS i stedet for MLST. Foreløpig er bruken av FGS forbeholdt de som har teknologien tilgjengelig og bioinformatikere som kan håndtere de store datamengdene, men etter hvert som bioinformatikk blir et mer utbredt fagfelt, vil FGS være en foretrukket teknologi for genotyping (27). Inntil videre vil imidlertid MLST-metoder som er nøye designet og som følger retningslinjer for validering av MLST, være en av de mest detaljerte genotypingsmetodene (27).
Utvikling av genetisk mangfold
Studier av det genetiske mangfoldet hos Borrelia-stammer og kartlegging av tilhørende epidemiologiske data (geografisk opprinnelse, vektor, vertsdyr), har gitt indikasjoner på at noen miljøfaktorer påvirker det genetiske mangfoldet mer enn andre. Geografisk opprinnelse og vektor-vertsdyr interaksjon, er de to determinantene som antas å være de viktigste påvirkningsfaktorene for evolusjon (figur 2) (8, 16). For å beskrive nye Borrelia-stammer brukes målgener som tidligere beskrevet: 16S rRNA, IGS, OSPs og utvalgte husholdningsgener kombinert i MLST- og MLSA-metoder. De ulike genene beskriver genetisk mangfold på forskjellige nivå.

Husholdningsgener og 16S rRNA er nyttige til genetisk karakteristikk og taksonomi. Artsspesifikke studier krever et høyere nivå av mutasjoner og det finner man i gener som IGS og OSPs. De ulike OSPs har forskjellig nivå av variasjon i ulike arter. Eksempelvis viser ospA et mindre genetisk mangfold hos B. afzelii-stammer og B. burgdorferi s.s.-stammer enn hos B. garinii-stammer. Studier av ospA har blant annet avdekket horisontal genoverføring mellom Borrelia-arter. Genet ospC er beskrevet som det av OSPs med høyeste grad av sekvensvariasjon, og er mye brukt i studier av patogen – vertsdyr interaksjon. Graden av det genetiske mangfoldet varierer mellom forskjellige geografiske områder, og utbredelsen av ulike Borrelia-arter ser ut til å være geografisk betinget. Blant annet viser prevalensstudier at de humanpatogene Borrelia-artene har ulik utbredelse på verdensbasis. I Europa finnes alle de patogene Borrelia-artene; B. afzelii, B. garinii, B. burgdorferi s.s. (og B. bavarensis) mens i USA regnes B. Burgdorferi s.s. for å være den eneste humanpatogene Borrelia-arten i B. burgdorferi s.l.-gruppen. I Asia finner man B. afzelii, B. garinii og B. bavarensis (8, 28, 29).
Flått fra slekten Ixodes er den vanligste vektoren for B. burgdorferi s.l. Flåttens manglende evne til å krysse store geografiske avstander gjør at den erverver patogener fra vertsdyr i et begrenset område, og utbredelsen av flåttbårne patogener avhenger til en viss grad av vertsdyrenes forflytning mellom geografiske områder. Landskapet og de ulike vertsdyrenes naturlige levested kan ha ulik påvirkning på det genetiske mangfoldet hos Borrelia-bakterier. Forurensning, ernæring, vann og naturlig stråling kan også påvirke (8, 16, 29).
Evolusjonsstudier har kartlagt slektskapsforholdet mellom Borrelia-stammer innsamlet på verdensbasis, og viser en klar sammenheng mellom geografisk opprinnelse og nukleotid-polymorfismer. Et stort genetisk mangfold blant analyserte B. afzelii-stammer settes i sammenheng med et begrenset trekkmønster hos vertsdyr som er reservoar for B. afzelii. Den viktigste gruppen vertsdyr for B. afzelii er smågnagere, som har territoriell tilknytning og dermed skaper geografisk tilknyttede B. afzelii-genotyper. Bare noen få B. afzelii-genotyper har blitt oppdaget på mer enn ett geografisk område. Dette skiller dem fra evolusjonære trekk hos for eksempel B. garinii-stammer, som har fugler som viktigste vertsdyr. Trekkfugler kan introdusere B. garinii til nye geografiske områder, noe som gir mulighet for spredning (28). Tilpasning til ulike vertsdyr har også en innvirkning på det genetiske mangfoldet. En spesifikk gruppe av B. garinii-stammer funnet i smågnagere har blitt omdøpt til B. bavarensis, ettersom genetiske egenskaper skiller dem fra B. garinii funnet i fugler. Denne assosiasjonen til vertsdyr tyder på at artsspesialisering og tilpasning til nye vertsdyr spiller en rolle i den genetiske utviklingen av Borrelia-stammer (7). Vertsdyrets komplementsystem vil kunne hindre Borrelia-infeksjon dersom Borrelia-stammen ikke er komplementær med vertsdyret. Gjennom blodmåltidet flåtten inntar fra vertsdyret, kan vertens komplementsystem potensielt ta livet av Borrelia-bakteriene som er inne i selve flåtten (16, 30).
Konklusjon
B. burgdorferi s.l. er et vektorbåret patogen som sirkulerer mellom vertsdyr og vektor, og denne parasittiske levemåten påvirker evolusjon og levemønster. Borrelia-genomet har noen spesielle genetiske egenskaper, og den store genetiske endringen og behovet for tilpasning til nye vertsdyr understreker behovet for detaljerte genotypingsmetoder som MLST og MLSA. Nettbaserte genotypings-databaser bidrar til en bedre forståelse av utviklingen av Borrelia-arter, og den rolle vektorer og vertsdyr spiller i evolusjonen og spredningen av bakterien. MLST- og MLSA-metoder er viktige verktøy for epidemiologiske studier av Borrelia, selv om det i fremtiden vil bli mer utstrakt bruk av fullgenom-sekvensering.